5. 快速开始
5.1. 生成 LAMMPS param.qeq 文件
在使用 ReaxFF 力场的 LAMMPS 模拟中,常需对原子电荷进行动态调整以保持体系电荷平衡。可以使用 fix qeq/reaxff 命令完成此操作,param.qeq 文本文件包含了每种元素的电荷平衡参数,该文件通常可从 ReaxFF 力场文件中提取。示例用法:
fix 1 all qeq/reaxff 1 0.0 10.0 1.0e-6 param.qeq
AutoRMA 提供从 ReaxFF 力场文件中提取电荷平衡参数并生成 LAMMPS 所需的 param.qeq 文件的功能。
步骤:
在菜单栏选择
Tools -> Extract ParamQeq File/工具 -> 提取电荷平衡参数文件,或点击工具栏中的相应按钮后打开操作面板;选择力场文件(纯文本格式,例如
ffield.reax)并填写输出文件路径;点击
Extract/提取完成提取。
备注
当前仅支持纯文本格式的力场文件,不支持 PDF/Word 等富文本格式文件。
5.2. 生成 LAMMPS data 文件
在 LAMMPS 中可通过 read_data 命令读取包含原子或分子系统初始配置的 data 文件,简化建模流程。
read_data reaxff.data
AutoRMA 提供从 Material Studio (MS) 导出的 CAR/MDF 文件生成 LAMMPS data 文件的功能,支持定制化处理原子类型、备注信息等内容。
步骤:
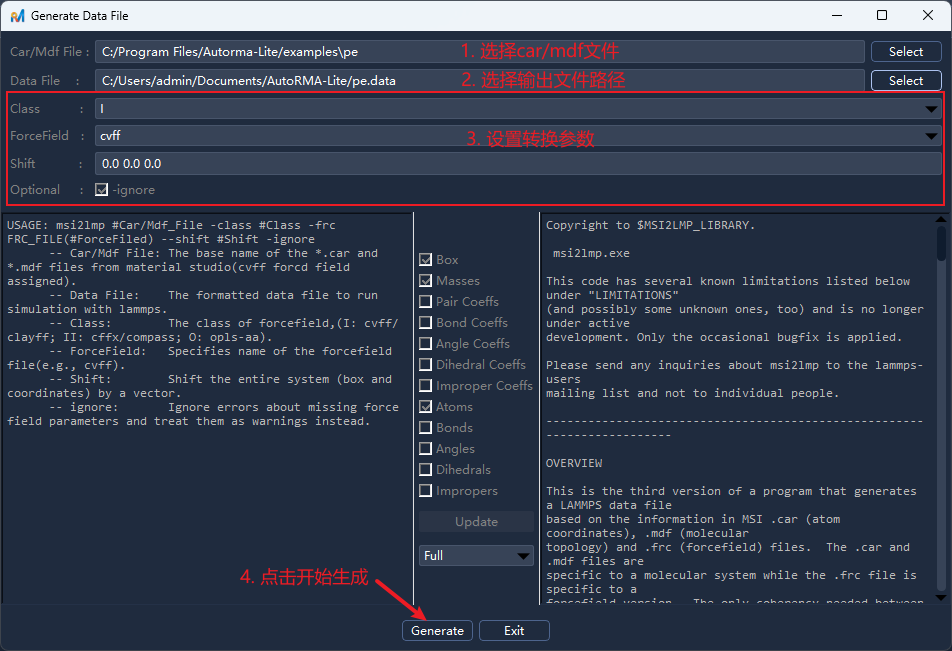
在菜单栏选择
Tools -> Generate Data File/工具 -> 生成Data文件,或点击工具栏按钮后打开操作面板;选择
CAR/MDF文件并填写输出文件路径,设置相关参数后点击Generate/生成;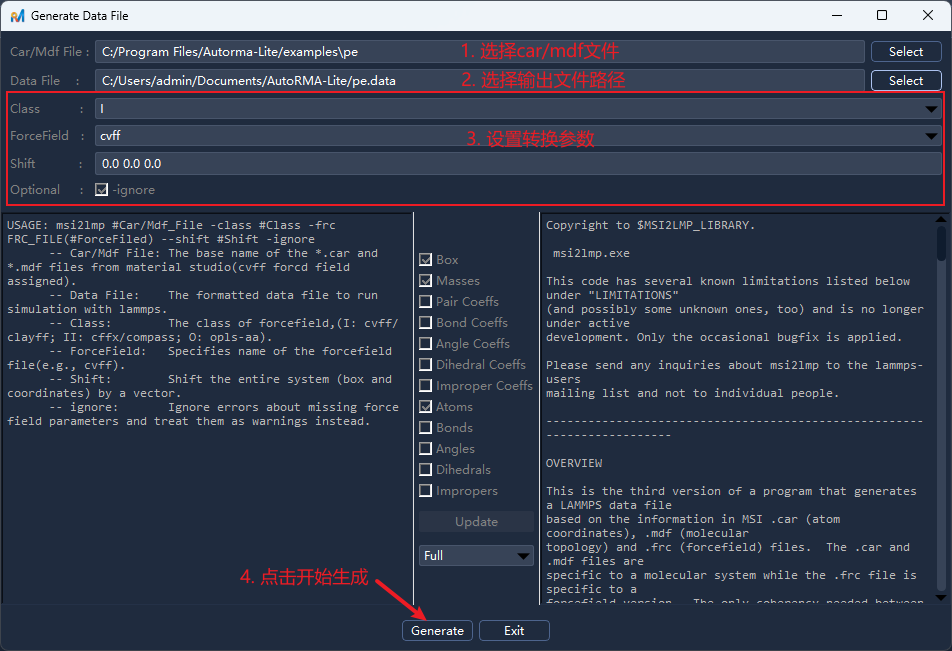
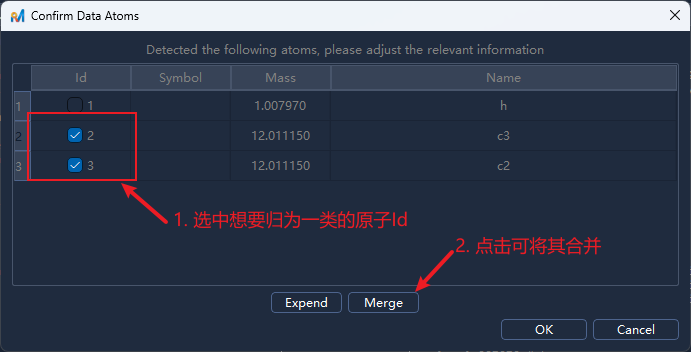
点击
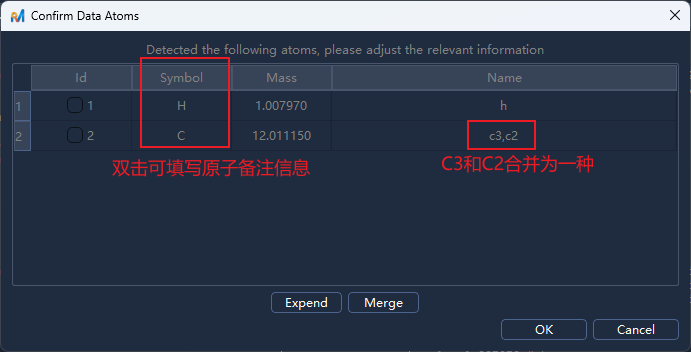
Generate/生成后会弹出原子信息确认面板(示例中存在 H、C2、C3 三类原子)。此时可进行以下操作:合并原子类型,如将 C2 和 C3 合并为 C,选中C2 C3原子后点击
Merge/合并,再点击确认即可调整原子顺序和原子id,双击原子id单元格进入编辑模式,输入新的id后按回车确认
添加文本信息,如在对应的Symbol单元格中添加元素符号(例如 C、H)方便后续查看和分析
确认无误后点击
OK/确认生成 data 文件,完成后的结果如下(左侧为 AutoRMA 处理后的结果,右侧为 LAMMPS-msi2lmp 原始输出)。可以看到 AutoRMA 生成的 data 文件已经按照 LAMMPS 的格式进行了重新组织,并且根据用户输入信息进行了调整,方便后续使用和分析。在信息确认面板中,勾选要包含的字段后点击
Update/更新可实时预览文件内容的变化,帮助用户确认最终输出格式若需修改原子类型模式(例如将从
Full切换为Charge或Atomic),在下拉选择框中进行选择,确认后点击面板底部的Generate/生成重新生成新的文件内容
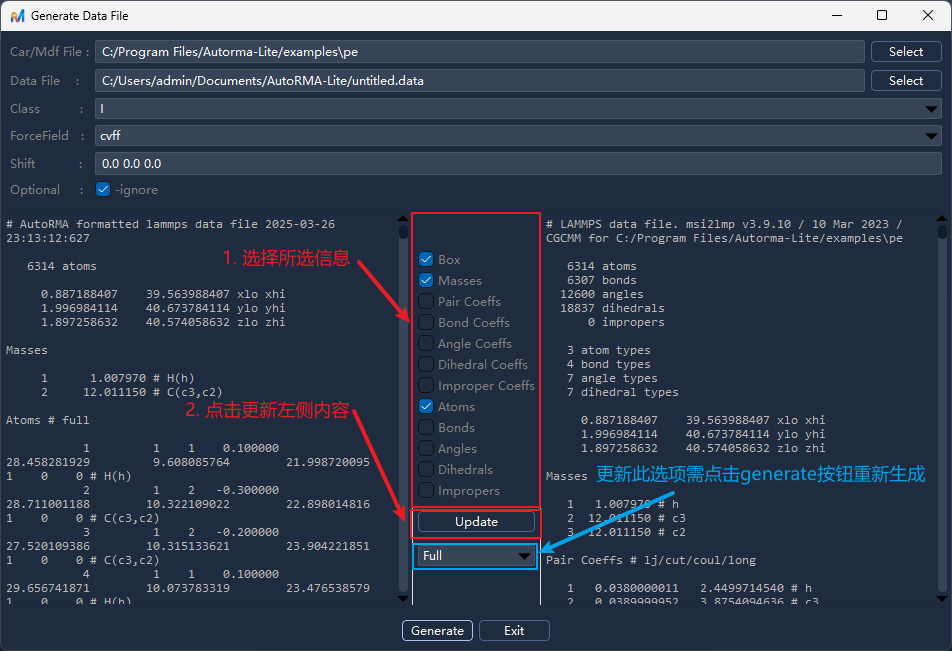
图 4.2.4: Data 文件结果展示
5.3. 创建 Species-Based/基于物种 项目
LAMMPS 可通过 fix reaxff/species 导出 species(物种/分子)文件,详细信息见 分子文件简介。AutoRMA 的核心功能之一是对物种文件进行分析。
步骤:
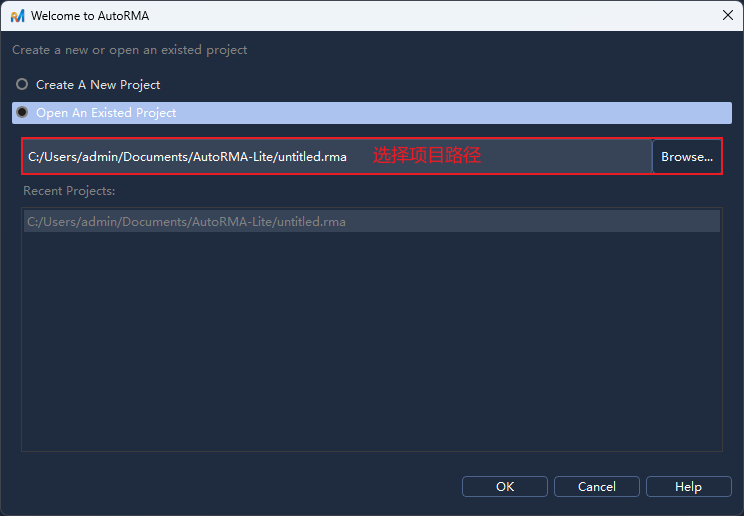
在启动软件后,选择新建项目(或在菜单栏选择
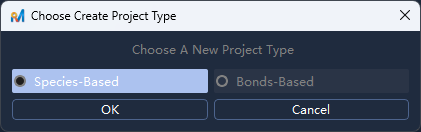
File -> New/文件 -> 新建),弹出文件对话框,新建一个后缀名.rma项目文件路径并确认;在弹出的
Choose Project Type/选择项目类型对话框中选择Species-Based/基于物种,点击OK/确定;
在
Create Species-Based New Project/创建基于物种的新项目面板中填写以下信息:参数
是否必填
说明
Species File/物种文件
必填
LAMMPS ReaxFF 输出的 species(*.out) 文件
Project Path/项目路径
必填
项目文件路径,后缀为
.rma,其同名文件夹将用于存储分析结果Atomic Symbol/原子符号
必填
参与模拟的元素符号,以空格分隔,例如
C H;顺序决定分子式书写顺序(如
C H则显示C2H4,H C则显示H4C2);
CPU Cores/CPU核数
可选
并行分析使用的 CPU 核心数,默认为系统总核心数减1,减1以避免耗尽资源造成设备卡顿
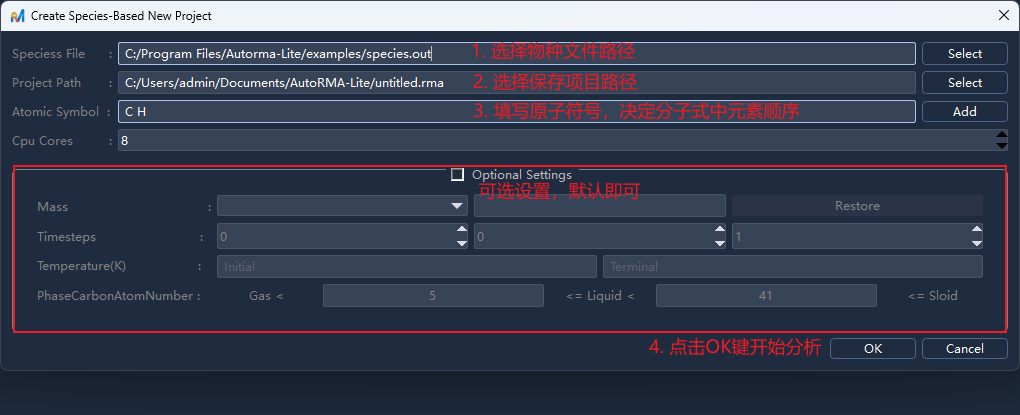
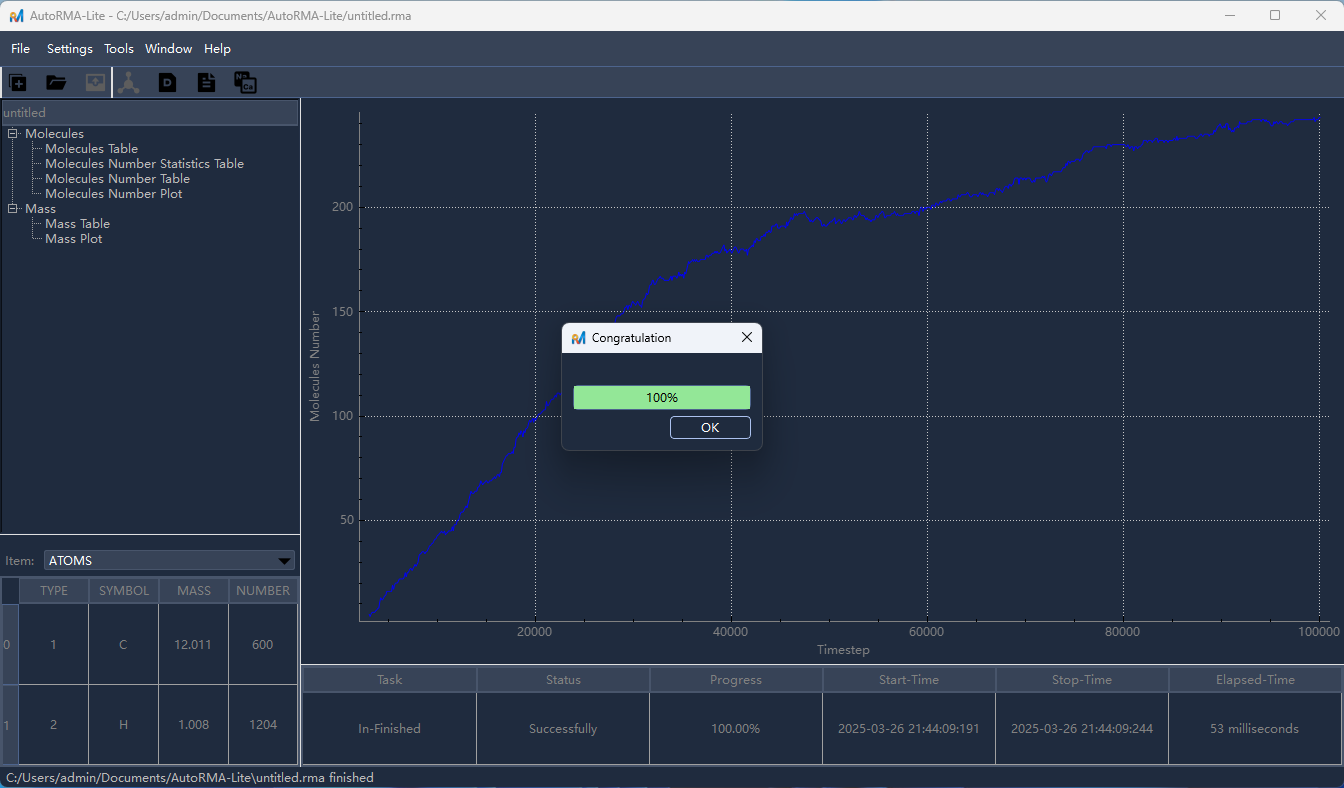
点击
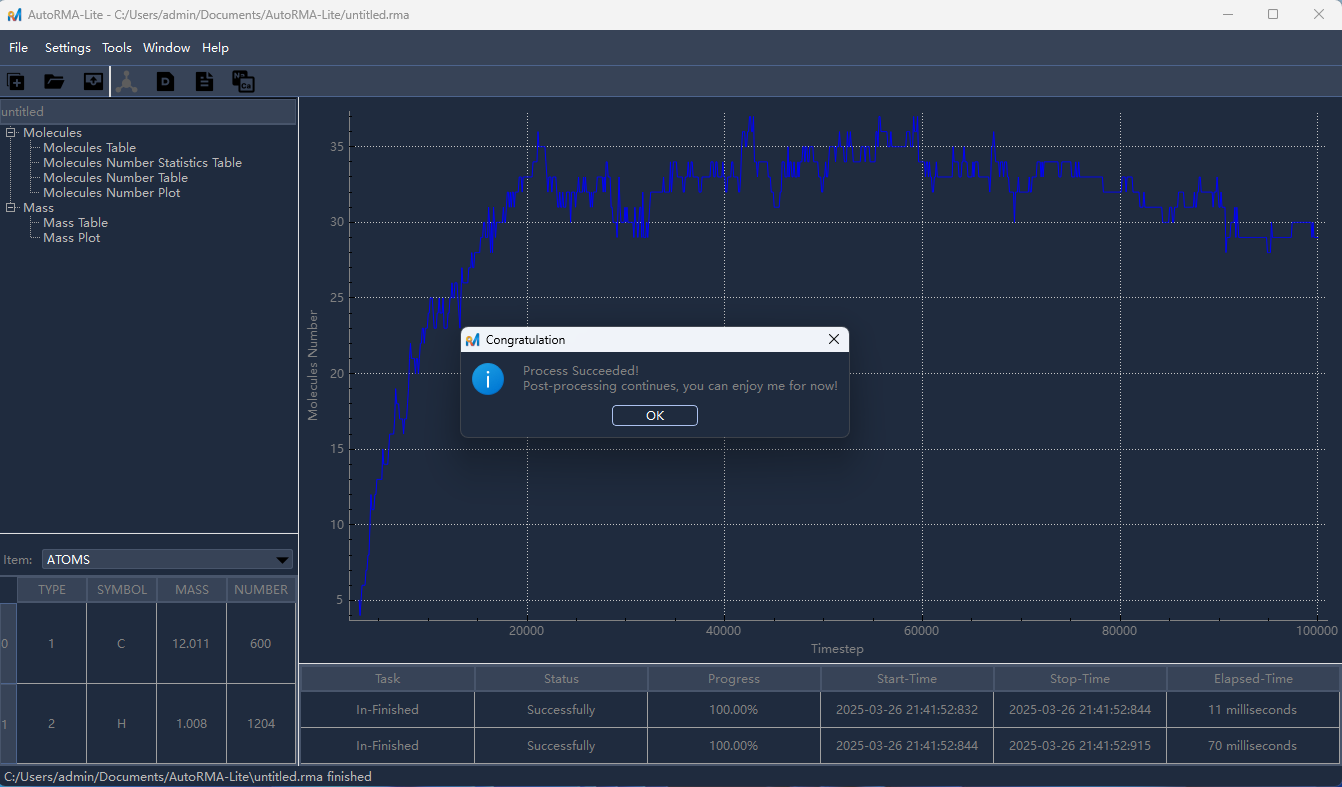
OK/确认开始分析,分析完成后,左侧目录树将显示分析结果条目,点击相应条目即可在右侧查看对应的分析结果。
5.4. 创建 Bonds-Based/基于键序 项目 (仅 AutoRMA-Full)
LAMMPS 可通过 fix reaxff/bonds 导出 bonds(键序)文件,详细信息见 键序文件简介。AutoRMA 的核心功能之一是对键序文件进行分析。
步骤: 操作面板与基于物种项目类似,以下不再展示实际操作界面。
在启动软件后,选择新建项目(或在菜单栏选择
File -> New/文件 -> 新建),弹出文件对话框,新建一个后缀名.rma项目文件路径并确认;在弹出的
Choose Project Type/选择项目类型对话框中选择Bonds-Based/基于键序,点击OK/确定;在
Create Bonds-Based New Project/创建基于键序的新项目面板中填写以下信息:参数
是否必填
说明
Bonds File/键序文件
必填
LAMMPS ReaxFF 输出的 bonds(*.reaxc) 文件
Trajs File/轨迹文件
必填
LAMMPS ReaxFF 输出的 trajs(*.lammpstrj) 文件
Project Path/项目路径
必填
项目文件路径,后缀为
.rma,其同名文件夹将用于存储分析结果Atomic Symbol/原子符号
必填
参与模拟的元素符号,以空格分隔,例如
C H;必须与 LAMMPS 输入脚本中的原子类型顺序一致,分子式书写格式与该顺序相同(如
C H则显示C2H4,H C则显示H4C2);可点击
Add/增加通过元素周期表面板选择;可点击
Details/详情逐元素配置原子质量与键数限制等信息。
CPU Cores/CPU核数
可选
并行分析使用的 CPU 核心数,默认为系统总核心数减1,减1以避免耗尽资源造成设备卡顿
点击
OK/确认开始分析,分析完成后,左侧目录树将显示分析结果条目,点击相应条目即可在右侧查看对应的分析结果。
5.5. 打开已有项目
在菜单栏选择
File -> Open/文件 -> 打开,或点击文件工具栏中的Open/打开按钮打开文件对话框;在弹出的文件对话框中选择以
.rma结尾的项目文件,点击Open/打开按钮打开项目;项目加载完成后,左侧目录树将显示分析结果条目,点击相应条目即可在右侧查看对应的分析结果。